

Adverse Experience Reporting
Case Report Form and Source Document
Managing and reporting adverse events – Key learning objectives
By the end of this session, you will be able to:
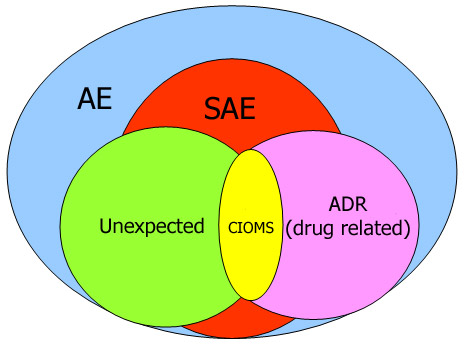
Adverse Event (AE)
ICH: AE
Serious Adverse Event (SAE) Definitions: ICH
Any adverse experience occurring at any dose that:
Differences between ‘serious’ and ‘severe’ (ICH)
The term ‘severe’ is used to describe the intensity (severity) of a special event (as in mild, moderate, severe myocardial infarction); the event itself, however, may be of relatively minor medical significance (such as severe headache).
This is not the same as ‘serious ’which is based on patient/ event outcome or action criteria usually associated with event that pose a threat to a patient’s life or functioning.
Seriousness (not severity) serves as a guide for defining regulatory reporting obligations.
What is NOT an AE
ADR and Unexpected ADR (ICH)
Adverse Drug Reaction (ADR)
Unexpected ADR
Documentation & Describing Adverse Events
Assessment of Causality (Relationship to the Test Drug)
Definitely Not Related
Probably Not Related
Possibly Related
Probably Related
Definitely Related
Investigator Reporting Requirements:ICH # 4.11
4.11.1 All SAEs should be reported immediately to the sponsor except for those SAEs that the protocol or other document (e.g., Investigator’s Brochure) identifies as not needing immediate reporting.
The immediate reports should be followed promptly by detailed, written reports.
The immediate and follow-up reports should identify subjects by unique code numbers assigned to the trial subjects.
The investigator should also comply with the applicable regulatory requirement(s) related to the reporting of unexpected serious adverse drug reactions to the regulatory authority(ies) and the IRB/ IEC.
4.11.2 AEs and/ or laboratory abnormalities identified in the protocol as critical to safety evaluations should be reported to sponsor according to the reporting requirements and within the time periods specified by the sponsor in the protocol.
4.11.3 For reported deaths, the investigator should supply the sponsor and the IRB/IEC with any additional requested information (e.g., autopsy report and terminal medical reports).
CIOMS
The Council for International Organizations of Medical Sciences
 Summary: Investigator Reporting Requirements
Summary: Investigator Reporting Requirements
Adverse Event Follow-Up Requirements Investigator responsibilities
Adverse Event Follow-Up Requirements
Possibly Sponsor actions:
Case studies
Case # 1
A 48-year-old white male subject with medical histories of renal stones, hypertension, diabetes and hypercholesterolemia was enrolled in protocol X. The subject received study drug on 10-Sep-2004. On 03-Nov-2005 the subject had an onset of kidney stones that required an overnight hospitalization (05-Nov-2005 to 06-Nov-2005) for a lithotripsy procedure, basket catch and urethral stent placement. The subject’s BUN and creatinine were elevated. There were no complications during surgery. No action was taken with study medication in response to the event. The subject was considered recovering from kidney stones. The investigator considered that there was not a reasonable possibility that the event was related to the study medication.
Case # 2
A 61-year-old Thai male subject with a history of chronic lumbar instability and cardiovascular condition was enrolled in study D. He began receiving run-in study drug on 08-Jul-2004. On 26-Aug-2004, he had prescheduled elective spinal fusion surgery and was discharged from the hospital on 14-Sep-2004. Several days later he developed a surgical site infection and on 23-Sep-2004 he was hospitalized with septicemia. His condition progressed to multiorgan failure. The patient was treated with metronidazole, folate, aspirin, hydrocortisone, and isotropic support. Study drug was stopped and he was discontinued from the study due to his deteriorating condition. On 03-Oct-2004, he died despite the extensive treatment. It was not known whether an autopsy would be performed.
Case # 3
A 71-year-old white male subject with medical history of high blood pressure and atrial fibrillation was enrolled in study Y. He started receiving study drug on 23-Nov-2005. On 16-Nov-2005, the subject’s hemoglobin was 16.7 g/dl (reference range was 13.1 to 16.5 g/dl). On 08-Dec-2005, his hemoglobin increased to 20.7 g/dl. He was diagnosed on the same day as having polycythemia. On 26-Jan-2006, the subject’s hemoglobin level was still high at 20.3 g/dl, and his hematocrit was also elevated. Because of the polycythemia, the subject underwent venesections on 27-Jan-2006, 30-Jan-2006, and 31-Jan-2006. The study drug was continued unchanged and the outcome of the event remains unknown as of the time of this report (03-Mar-2006). The investigator considered that there was a reasonable possibility that polycythemia was related to the study drug.
Case Report Form and Source Document
Managing and reporting adverse events – Key learning objectives
By the end of this session, you will be able to:
- Define and classify an adverse event;
- Differentiate between serious and severe adverse events;
- Evaluate the causal relationship between an adverse event and a drug;
- Specify the investigator’s requirements for managing and reporting adverse events;
- Discuss practical consideration for soliciting and capturing adverse event data
Adverse Event (AE)
ICH: AE
- An AE is any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product that does not necessarily have a causal relationship with this treatment. An AE can therefore be:
- - any unfavorable and unintended sign (including an abnormal laboratory finding)
- symptom, or disease temporally associated with the use of a medicinal (investigational) product whether or not related to the medicinal (investigational) product
Serious Adverse Event (SAE) Definitions: ICH
Any adverse experience occurring at any dose that:
- *Results in death
- *Is life-threatening
- *Results in persistent or significant disability/incapacity
- *Results in hospitalization or prolongation of existing hospitalization
- *Is a congenital anomaly/birth defect
- Other important medical events that may not results in death, not be life-threatening, or not require hospitalization may be considered a serious adverse experience when, based on appropriate medical judgment, the event may jeopardize the patient and may require medical or surgical intervention to prevent one of the starred (*) outcomes listed previously
Differences between ‘serious’ and ‘severe’ (ICH)
The term ‘severe’ is used to describe the intensity (severity) of a special event (as in mild, moderate, severe myocardial infarction); the event itself, however, may be of relatively minor medical significance (such as severe headache).
This is not the same as ‘serious ’which is based on patient/ event outcome or action criteria usually associated with event that pose a threat to a patient’s life or functioning.
Seriousness (not severity) serves as a guide for defining regulatory reporting obligations.
What is NOT an AE
- Pre-existing conditions
- - Complete medical history is essential for accurate reporting of AEs
- However, a worsening (frequency or intensity) of a pre-existing condition is an AE
- Procedures
- - However, underlying condition (reason for the procedure) is the AE
- Elective procedures and their underlying causes
ADR and Unexpected ADR (ICH)
Adverse Drug Reaction (ADR)
- All noxious and unintended response to a medicinal product related to any dose should be considered adverse drug reactions.
- “response to a medicinal product” means that the causal relationship between a medicinal product and an adverse event is at least a reasonable possibility, i.e., the relationship can not be ruled out
Unexpected ADR
- An adverse reaction, the nature or severity of which is not consistent with the applicable product information (e.g., Investigator’s Brochure for an unapproved investigational medicinal product).
Documentation & Describing Adverse Events
- Reporter name, source type (physician or others)
- Subject data subject number, sex, age, medical history
- Subject’s drug data study drugs and concomitant drugs; daily dose, route of administration, indication, start and stop date
- Adverse event data AE term, onset and cessation date or duration, severity, seriousness, outcome, action taken as a result of AE, de-challenge, re-challenge, AE narrative (description of the event), investigator’s assessment of causal relationship, If death, provide cause of death
Assessment of Causality (Relationship to the Test Drug)
- Made by the qualified investigator, according to his/ her best clinical judgment
- 1. Definitely Not - No relationship
2. Probably Not - The relationship is not probable
3. Possibly - The relationship could exist
4. Probably - The relationship is probable
5. Definitely - The relationship is indisputable
Definitely Not Related
- The patient did not received the test drug,
- OR
- Temporal sequence of the AE onset relative to administration of the test drug is not feasible,
- OR
- There is another obvious cause of the AE (e.g., concurrent medical condition, disease under study (protocol indication), concomitant medications, study procedures)
Probably Not Related
- There is evidence of exposure to the test drug
- There is another more likely cause of the AE
- Dechallenge (if performed) is negative or ambiguous
- Rechallenge (if performed) is negative or ambiguous
Possibly Related
- There is evidence of exposure to the test drug
- The temporal sequence of the AE onset relative the administration of the test drug is reasonable
- The AE could have been due to another equally likely cause
- Dechallenge (if performed) is positive
Probably Related
- There is evidence of exposure to the test drug
- The temporal sequence of the AE onset relative the administration of the test drug is reasonable
- The AE is more likely explained by the test drug than by another cause
- Dechallenge (if performed) is positive
Definitely Related
- There is evidence of exposure to the test drug
- The temporal sequence of the AE onset relative the administration of the test drug is reasonable
- The AE is more likely explained by the test drug than by another cause
- Dechallenge is positive
- Rechallenge (if feasible) is positive
- The AE shows a pattern consistent with previous knowledge of the test drug or test drug class
Investigator Reporting Requirements:ICH # 4.11
4.11.1 All SAEs should be reported immediately to the sponsor except for those SAEs that the protocol or other document (e.g., Investigator’s Brochure) identifies as not needing immediate reporting.
The immediate reports should be followed promptly by detailed, written reports.
The immediate and follow-up reports should identify subjects by unique code numbers assigned to the trial subjects.
The investigator should also comply with the applicable regulatory requirement(s) related to the reporting of unexpected serious adverse drug reactions to the regulatory authority(ies) and the IRB/ IEC.
4.11.2 AEs and/ or laboratory abnormalities identified in the protocol as critical to safety evaluations should be reported to sponsor according to the reporting requirements and within the time periods specified by the sponsor in the protocol.
4.11.3 For reported deaths, the investigator should supply the sponsor and the IRB/IEC with any additional requested information (e.g., autopsy report and terminal medical reports).
CIOMS
The Council for International Organizations of Medical Sciences
- Established in 1949 by WHO and UNESCO
- CIOMS working groups prepared their reports and published their recommendations:
- - CIOMS I. 1990 International Reporting of Adverse Drug Reactions
- The most valuable outcome of the working group of CIOMS I was the introduction of the “CIOMS I reporting form” for standardized international reporting of individual cases of serious, unexpected adverse drug reactions (drug related).
- Record all adverse events (SAE and non-SAE) on CRFs (Case Report Forms)
- Report SAE to Sponsor immediately
- Report serious, unexpected ADR (CIOMS report) to regulatory authority and IRB/ IEC
- For death report, supply requested information to sponsor and IRB/ IEC
Adverse Event Follow-Up Requirements Investigator responsibilities
- Regardless of the specific study requirements, the investigator is responsible for personally:
- Treating, or overseeing the treatment of the AE;
- Following the subject until the AE resolves
- None
- Increase surveillance of the subject;
- Provide an antidote/ counteractive measures;
- Change the dose or dosing regimen;
- Discontinue study drug administration;
- Some combination of the above
Adverse Event Follow-Up Requirements
Possibly Sponsor actions:
- Confirm that the site has notified the EC;
- Notify appropriate regulatory agencies;
- Advise other Investigators participating in the study;
- Revise/ modify protocols and informed consents;
- Temporarily suspend the study;
- Discontinue the study;
- Temporarily suspend or permanently stop the drug development
- Revise/ modify protocols and informed consents;
- Temporarily suspend the study;
- Discontinue the study
Case studies
Case # 1
A 48-year-old white male subject with medical histories of renal stones, hypertension, diabetes and hypercholesterolemia was enrolled in protocol X. The subject received study drug on 10-Sep-2004. On 03-Nov-2005 the subject had an onset of kidney stones that required an overnight hospitalization (05-Nov-2005 to 06-Nov-2005) for a lithotripsy procedure, basket catch and urethral stent placement. The subject’s BUN and creatinine were elevated. There were no complications during surgery. No action was taken with study medication in response to the event. The subject was considered recovering from kidney stones. The investigator considered that there was not a reasonable possibility that the event was related to the study medication.
- Q1: What is the AE (s)?
Q2: If it is considered serious, what is the serious criterion?
Case # 2
A 61-year-old Thai male subject with a history of chronic lumbar instability and cardiovascular condition was enrolled in study D. He began receiving run-in study drug on 08-Jul-2004. On 26-Aug-2004, he had prescheduled elective spinal fusion surgery and was discharged from the hospital on 14-Sep-2004. Several days later he developed a surgical site infection and on 23-Sep-2004 he was hospitalized with septicemia. His condition progressed to multiorgan failure. The patient was treated with metronidazole, folate, aspirin, hydrocortisone, and isotropic support. Study drug was stopped and he was discontinued from the study due to his deteriorating condition. On 03-Oct-2004, he died despite the extensive treatment. It was not known whether an autopsy would be performed.
- Q1: What is the AE(s)?
Q2: If it is considered serious, what is the serious criterion?
Q3: How would you assess the causality of the AE to the test drug?
Case # 3
A 71-year-old white male subject with medical history of high blood pressure and atrial fibrillation was enrolled in study Y. He started receiving study drug on 23-Nov-2005. On 16-Nov-2005, the subject’s hemoglobin was 16.7 g/dl (reference range was 13.1 to 16.5 g/dl). On 08-Dec-2005, his hemoglobin increased to 20.7 g/dl. He was diagnosed on the same day as having polycythemia. On 26-Jan-2006, the subject’s hemoglobin level was still high at 20.3 g/dl, and his hematocrit was also elevated. Because of the polycythemia, the subject underwent venesections on 27-Jan-2006, 30-Jan-2006, and 31-Jan-2006. The study drug was continued unchanged and the outcome of the event remains unknown as of the time of this report (03-Mar-2006). The investigator considered that there was a reasonable possibility that polycythemia was related to the study drug.
- Q1: If polycythemia was considered a SAE, what is the serious criterion?
Q2: How would you fill in the laboratory page of this AE?
Q3: What are investigator’s possible actions to this case?